Iako je ljudska potraga za transformacijom materijala bila složena i beskonačna tokom istorije — od pokušaja alhemičara da stvore zlato iz običnih supstanci do savremenih pristupa — najnovije istraživanje donosi značajan iskorak. Grupa naučnika sa MIT-a, predvođena profesorom Ju Li, razvila je metod koji koristi mašinsko učenje za značajno ubrzanje i unapređenje procesa predviđanja svojstava materijala, pomičući granice znanja u oblasti računarske hemije.
U tradicionalnoj računarskoj hemiji, većina metoda za predviđanje molekularnih sistema oslanja se na teoriju funkcionalne gustine (DFT), koja pruža kvantno-mehanički pristup za određivanje energije molekula. Iako je DFT metoda bila vrlo uspešna, njena tačnost može biti neujednačena, a koristi se uglavnom za određivanje najniže energije molekularnih sistema.
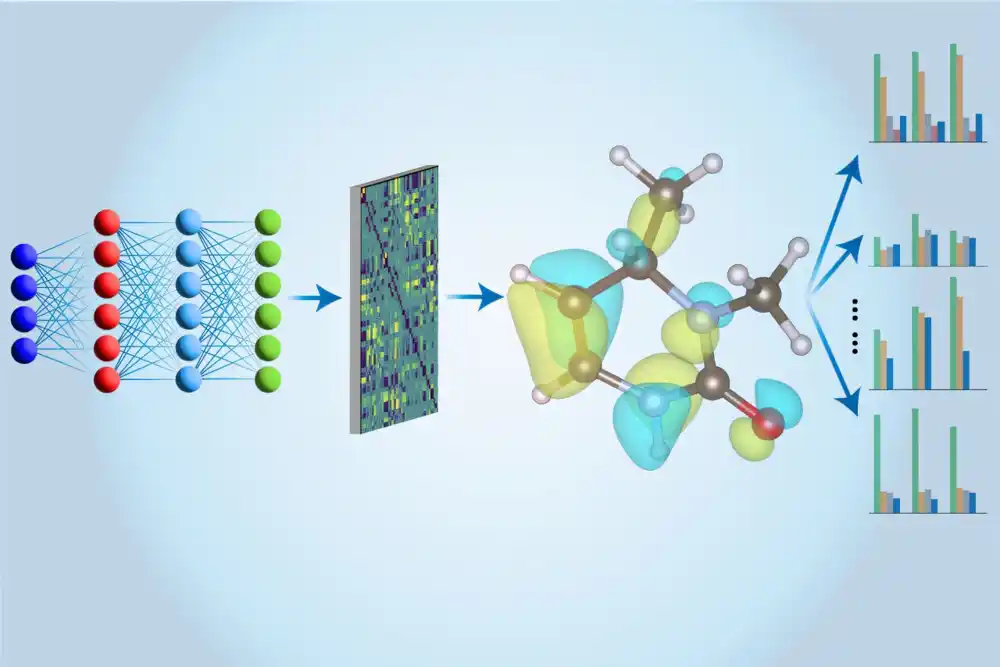
Međutim, nova tehnika, teorija spregnutih klastera (CCSD(T)), nudi daleko preciznije rezultate, ali ima velike računarske troškove i nije skalabilna za veće sisteme. Kako bi rešili ovaj problem, Li i njegov tim primenili su mašinsko učenje, koristeći neuronske mreže za preciznu aproksimaciju rezultata proračuna CCSD(T). Ovaj pristup omogućava značajna ubrzanja proračuna, jer neuronska mreža, nakon obuke na manjim molekulima, može brzo izvoditi iste proračune, dok istovremeno izdvaja dodatne informacije o molekulama, kao što su njihov dipolni moment ili optical gap.
Jedna od ključnih inovacija tima je pristup više zadataka, u kojem se koristi jedna neuronska mreža za procenu različitih svojstava molekula, što je mnogo efikasnije od prethodnih metoda koje su koristile različite modele za svako svojstvo. Ovaj model, poznat kao MEHnet, koristi naprednu mrežnu arhitekturu koja uključuje fizičke principe, omogućavajući ne samo proračun osnovnih svojstava već i svojstava uzbuđenih stanja molekula.
Prednosti novog pristupa:
- Veća preciznost: Model je pokazao veću tačnost u poređenju sa DFT-om, posebno u testiranjima sa poznatim ugljovodoničkim molekulima.
- Brža računanja: Proračuni se obavljaju mnogo brže, omogućavajući analizu većih molekula sa hiljadama atoma, što je ranije bilo gotovo nemoguće.
- Veća skalabilnost: Iako je proračun sa CCSD(T) metodom bio ograničen na molekule sa manjim brojem atoma, ovaj model može obraditi sisteme sa stotinama i potencijalno hiljadama atoma.
Potencijal za industriju i nauku: Jedan od najuzbudljivijih aspekata ovog istraživanja je njegov potencijal za brzi molekularni screening visoke propusnosti, što omogućava istraživačima da efikasnije identifikuju nove materijale sa željenim svojstvima. Ovo može biti od presudne važnosti za razvoj novih lekova, polimera, i baterija sa boljim performansama.
Gledajući napred, Li i njegov tim planiraju da prošire ovu metodologiju na teže elemente i veće sisteme, što bi moglo doneti revoluciju u oblasti dizajniranja materijala. Kako bi postigli ovaj cilj, ambicija istraživača je da obuhvate ceo periodni sistem sa visokom preciznošću, ali uz niže računarske troškove, što bi moglo imati široke primene u nauci o materijalima, biologiji i hemiji.
Kao što je primetio specijalista za otkrivanje materijala Ćiang Žu sa Univerziteta Severne Karoline, sinergija između računarske hemije i dubokog učenja ima ogroman potencijal da unapredi istraživanja u ovoj oblasti, omogućavajući preciznije, skalabilnije i brže metode za dizajniranje novih materijala i molekula.


