Nedavna studija objavljena u Nature Metabolism otkrila je patogeni mehanizam koji leži u osnovi retkog pedijatrijskog neurodegenerativnog poremećaja poznatog kao sindrom neurodegeneracije povezane sa proteinom mitohondrijalne enoil reduktaze (MEPAN).
Studiju su vodili dr Hugo J. Bellen, istaknuti profesor usluga na Medicinskom koledžu Bejlor i predsedavajući za neurogenetiku na Institutu za neurološka istraživanja Jan i Dan Dankan u Dečjoj bolnici u Teksasu (Duncan NRI), i dr Debdip Duta, postdoktorski saradnik u laboratoriji Bellen.
Tim Duncan NRI je otkrio da kod pacijenata i životinjskih modela ovog poremećaja, veliki broj neurona umire zbog prekomerne akumulacije ceramida i neispravnog metabolizma gvožđa, što je rezultat poremećaja u sintezi mitohondrijalnih masnih kiselina. On je prvi koji obezbeđuje mehaničku vezu između poremećaja u sintezi mitohondrijalnih masnih kiselina, metabolizma gvožđa i ceramida i neurodegeneracije.
Masne kiseline su osnovni gradivni blokovi složenih lipida u našem telu. U većini višećelijskih organizama, uključujući ljude, glavni deo masnih kiselina se sintetiše u citoplazmi, želatinoznoj tečnosti koja ispunjava većinu ćelija. Krajem osamdesetih godina prošlog veka otkriveno je da se mali deo masnih kiselina takođe sintetiše u mitohondrijima, koji deluju kao generatori energije ćelije.
Godine 2016, mutacije u genu mitohondrijalne enoil coA-reduktaze (MECR) identifikovane su kao uzrok MEPAN sindroma, retkog neurološkog stanja koje karakterišu progresivni motorički problemi, kao što su distonija, problemi sa govorom i gubitak vida, što je na kraju dovelo do slepilo. MECR gen kodira enzim koji katalizuje poslednji korak u sintezi mitohondrijalnih masnih kiselina, ali se vrlo malo znalo o tačnom mehanizmu kojim je poremećaj ovog gena uticao na stabilnost i funkciju neurona.
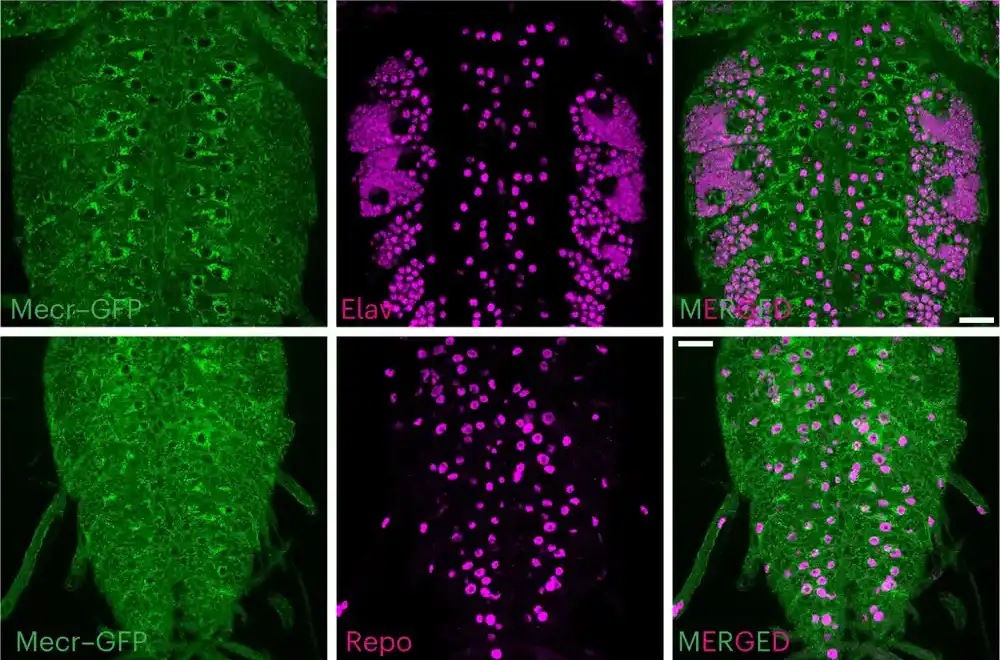
„Da bismo dešifrovali koji biološki procesi i putevi krenu po zlu kada je MECR gen poremećen, koristili smo CRISPR tehnologiju da izbrišemo ovaj gen kod voćnih mušica“, rekao je dr Belen, koji je takođe profesor razvojne biologije u Bejloru.
Videli su da muve kojima su nedostajale obe kopije mecr gena nisu preživele, dok je prisustvo jedne netaknute kopije muve ili ljudske verzije ovog gena bilo dovoljno za preživljavanje. Preživeo je samo mali deo muva koje su ispoljile mutantnu (koja izazivaju) varijantu – što ukazuje da je smrtnost zaista bila posledica gubitka funkcije MECR gena. Slično pacijentima sa MEPAN-om, muve koje nose mutantnu verziju gena fli mecr pokazale su progresivne probleme mobilnosti povezane sa uzrastom, smanjenu neuronsku aktivnost u neuronima mrežnjače i druge znake neurodegeneracije.
„Zanimljivo je da smo otkrili da su mitohondrije u mecr mutantima i ćelijama fibroblasta pacijenata MEPAN strukturno i funkcionalno abnormalne“, rekao je dr Duta, prvi autor studije. „Dalje, lipidomske i druge analize su otkrile da iako su nivoi većine fosfolipida ostali nepromenjeni, došlo je do povećanja sfingolipida kao što su ceramidi i drugih metabolita kao što je gvožđe. U poređenju sa drugim ćelijama, neuroni troše mnogo ćelijske energije i tako , očekuje se da će ove promene narušiti funkciju neurona kod mecr mutanata i MEPAN pacijenata.“
„Najviše nas je zaintrigiralo da vidimo povećane nivoe ceramida i defekte u metabolizmu gvožđa u modelima MEPAN sindroma i ćelijama dobijenim od ovih pacijenata“, rekao je Bellen.
„Nekoliko prethodnih studija iz naše laboratorije i drugih prijavilo je uporedivo povećanje ovih metabolita kod pacijenata i modela muva drugih progresivnih neurodegenerativnih poremećaja kao što su Fredreichova ataksija, infantilna neuroaksonalna distrofija, Gošerova bolest i Parkinsonova bolest.
„Ovaj rad još jednom naglašava kritičnu važnost održavanja tačnih nivoa mitohondrijalnih masnih kiselina, ceramida i gvožđa kako bi se sprečio prevremeni gubitak neurona. Nadamo se da će rezultati ove studije unaprediti napore u razvoju lekova za pacijente sa MEPAN sindromom i povezani neurodegenerativni poremećaji“.