Objavljivanjem platformi kao što su DALL-E 2 i Midjournei, difuzioni generativni modeli su postigli popularnost u mejnstrimu, zahvaljujući svojoj sposobnosti da generišu niz apsurdnih, zadivljujućih i često dostojnih slika iz tekstualnih upita poput „plišanih medvedića koji rade na novim Istraživanje veštačke inteligencije na mesecu 1980-ih.“
Ali tim istraživača na MIT-ovoj klinici Abdul Latif Jameel za mašinsko učenje u zdravlju (Jameel Clinic) misli da bi moglo biti više od širenja generativnih modela od pukog stvaranja nadrealnih slika – oni bi mogli da ubrzaju razvoj novih lekova i smanje verovatnoću štetne strane efekti.
Rad koji predstavlja ovaj novi molekularni model spajanja, nazvan DiffDock, biće predstavljen na 11. međunarodnoj konferenciji o reprezentacijama učenja. Jedinstveni pristup ovog modela računarskom dizajnu lekova je promena paradigme u odnosu na trenutne najsavremenije alate koje koristi većina farmaceutskih kompanija, što predstavlja veliku priliku za reviziju tradicionalnog procesa razvoja lekova.
Lekovi obično funkcionišu u interakciji sa proteinima koji čine naša tela, ili proteinima bakterija i virusa. Molekularno spajanje je razvijeno da bi se stekao uvid u ove interakcije predviđanjem atomskih 3D koordinata sa kojima bi se ligand (tj. molekul leka) i protein mogli povezati.
Dok je molekularno spajanje dovelo do uspešne identifikacije lekova koji sada leče HIV i rak, pri čemu svaki lek u proseku ima deceniju vremena razvoja i 90 odsto kandidata za lek ne uspeva u skupim kliničkim ispitivanjima (većina studija procenjuje da su prosečni troškovi razvoja lekova oko milijardu dolara na preko 2 milijarde dolara po leku), nije ni čudo što istraživači traže brže i efikasnije načine da probiju potencijalne molekule leka.
Trenutno, većina molekularnih alata za pristajanje koji se koriste za in-silico dizajn lekova uzimaju pristup „uzorkovanje i bodovanje“, tražeći „pozu“ liganda koja najbolje odgovara proteinskom džepu. Ovaj dugotrajni proces procenjuje veliki broj različitih poza, a zatim ih ocenjuje na osnovu toga koliko se dobro ligand vezuje za protein.
U prethodnim rešenjima za duboko učenje, molekularno spajanje se tretira kao problem regresije. Drugim rečima, „pretpostavlja se da imate jednu metu za koju pokušavate da optimizujete i da postoji samo jedan tačan odgovor“, kaže Gabriele Corso, koautor i dr MIT na drugoj godini. student elektrotehnike i računarstva koji je podružnica MIT Laboratorije za računarske nauke i veštačku inteligenciju (CSAIL).
„Kod generativnog modeliranja, pretpostavljate da postoji distribucija mogućih odgovora – ovo je kritično u prisustvu neizvesnosti.“
„Umesto jednog predviđanja kao ranije, sada dozvoljavate da se predvidi više poza, i svaka sa različitom verovatnoćom“, dodaje Hanes Štark, koautor i doktorat na MIT-u prve godine. student elektrotehnike i računarstva koji je podružnica MIT Laboratorije za računarske nauke i veštačku inteligenciju (CSAIL). Kao rezultat toga, model ne mora da pravi kompromise u pokušaju da dođe do jednog zaključka, što može biti recept za neuspeh.
Da bismo razumeli kako funkcionišu generativni modeli difuzije, korisno je objasniti ih na osnovu modela difuzije koji generišu sliku. Ovde, difuzioni modeli postepeno dodaju nasumični šum na 2D sliku kroz niz koraka, uništavajući podatke na slici sve dok ne postanu ništa drugo do zrnasta statika. Neuronska mreža se zatim obučava da povrati originalnu sliku obrnutim procesom buke. Model tada može da generiše nove podatke počevši od nasumične konfiguracije i iterativno uklanjajući šum.
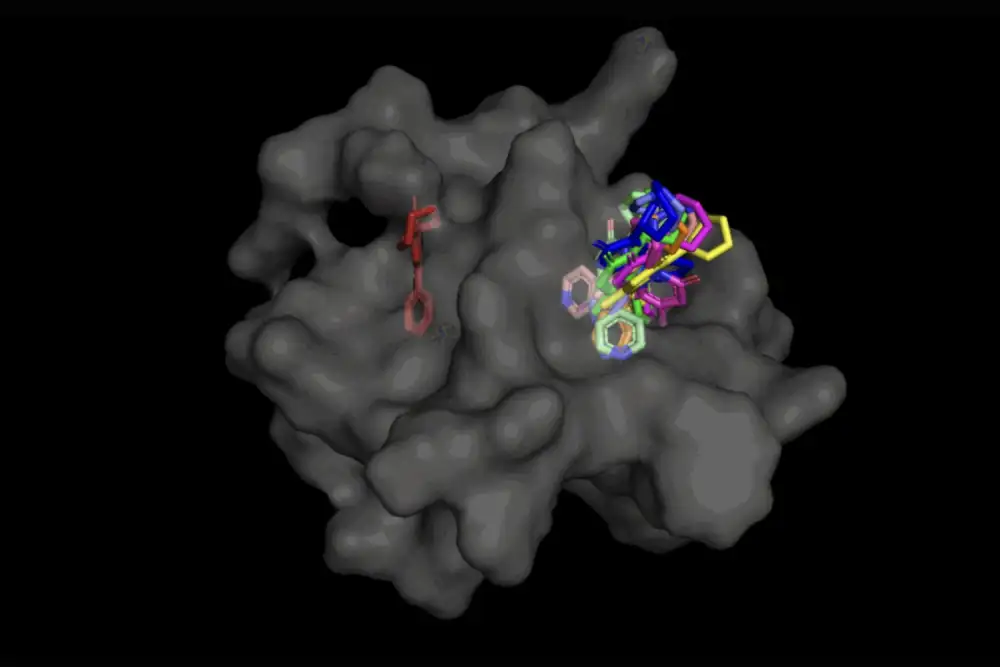
U slučaju DiffDock-a, nakon što je obučen za različite poze liganda i proteina, model je u stanju da uspešno identifikuje višestruka mesta vezivanja na proteinima sa kojima se nikada ranije nije susreo. Umesto da generiše nove podatke o slici, on generiše nove 3D koordinate koje pomažu ligandu da pronađe potencijalne uglove koji bi mu omogućili da stane u proteinski džep.
Ovaj pristup „slepog pristajanja“ stvara nove mogućnosti da se iskoristi AlphaFold 2 (2020), DeepMind-ov čuveni AI model za preklapanje proteina. Od početnog izdanja AlphaFold-a 1 2018. godine, u istraživačkoj zajednici vlada veliko uzbuđenje zbog potencijala AlphaFold-ovih kompjuterski presavijenih proteinskih struktura da pomognu u identifikaciji novih mehanizama delovanja lekova.
Ali najsavremeniji alati za molekularno pristajanje tek treba da pokažu da je njihov učinak u vezivanju liganada za računarski predviđene strukture bolji od slučajnog slučaja.
Ne samo da je DiffDock znatno tačniji od prethodnih pristupa tradicionalnim referentnim vrednostima za pristajanje, zahvaljujući svojoj sposobnosti da razmišlja na višoj skali i implicitno modelira deo fleksibilnosti proteina, DiffDock održava visoke performanse, čak i kada drugi modeli pristajanja počinju da ne uspevaju.
U realističnijem scenariju koji uključuje upotrebu računarski generisanih nevezanih proteinskih struktura, DiffDock postavlja 22 procenta svojih predviđanja unutar 2 angstroma (široko se smatra pragom za tačnu pozu, 1A odgovara jednoj preko 10 milijardi metara), više nego dvostruko drugi modeli za pristajanje jedva lebde preko 10 procenata za neke i padaju čak na 1,7 procenata.
Ova poboljšanja stvaraju novi pejzaž mogućnosti za biološka istraživanja i otkrivanje lekova. Na primer, mnogi lekovi se pronalaze putem procesa poznatog kao fenotipski skrining, u kome istraživači posmatraju efekte datog leka na bolest, a da ne znaju na koje proteine lek deluje.
Otkrivanje mehanizma delovanja leka je tada ključno za razumevanje kako se lek može poboljšati i njegovih potencijalnih neželjenih efekata. Ovaj proces, poznat kao „obrnuti skrining“, može biti izuzetno izazovan i skup, ali kombinacija tehnika savijanja proteina i DiffDock-a može omogućiti izvođenje velikog dela procesa u silikonu, omogućavajući da se identifikuju potencijalni neželjeni efekti „van cilja“. rano pre početka kliničkih ispitivanja.
„DiffDock čini identifikaciju cilja leka mnogo mogućim. Ranije su morali da se rade naporni i skupi eksperimenti (meseci do godine) sa svakim proteinom da bi se definisalo pristajanje leka. Ali sada se može pregledati mnogo proteina i izvršiti trijaža praktično za jedan dan “, kaže Tim Peterson, docent na Medicinskom fakultetu Univerziteta Vašington u St. Peterson je koristio DiffDock da okarakteriše mehanizam delovanja novog kandidata za lek koji leči bolesti povezane sa starenjem u nedavnom radu.
„Postoji veoma „sudbina voli ironiju“ aspekt da se Eroomov zakon — da otkrivanje lekova traje duže i košta više novca svake godine — rešava njegovim istoimenim Murovim zakonom — da računari svake godine postaju brži i jeftiniji — koristeći alate kao što je DiffDock .“


